



Введение
Множественная лекарственная устойчивость (МЛУ) – это невосприимчивость клеток или организма одновременно к целому ряду лекарственных препаратов разного химического строения и с разным механизмом действия. Она определяется как снижение чувствительности до такой степени, что клетки способны размножаться при воздействии на них препарата в критической или более высокой концентрации. Феномен МЛУ имеет важное клиническое значение, поскольку представляет собой серьезное препятствие на пути успешного лечения многих заболеваний, в том числе злокачественных опухолей. Развитие МЛУ к используемым лекарственным препаратам является одним из проявлений фундаментального биологического свойства всех живых организмов – приспособления к изменениям условий внешней среды. Исследования последних лет показали, что молекулярные механизмы МЛУ множественны, и лекарственная устойчивость может определяться включением различных биологических систем, характеризующих разные этапы осуществления токсического действия химиопрепарата – от ограничения накопления лекарства внутри клетки до отмены программы гибели клеток, индуцируемой веществом. Нередко в клетке включается несколько защитных механизмов, однако чаще всего преобладает какой-то один механизм. Наиболее изученными механизмами, клиническая значимость которых при определенных формах новообразований установлена, являются: активация трансмембранных транспортных белков, выводящих различные вещества из клетки (в частности, Р-гликопротеина – Pgp); активация ферментов системы глутатиона, детоксифицирующей препараты; изменения генов и белков, контролирующих апоптоз и выживаемость клеток (Ставровская, 2000; Анисимов и др., 2003; Volkova et al., 2012). Существует тесная взаимосвязь количественных изменений клеточной популяции и изменений их биологических свойств, одним из которых является лекарственная устойчивость. В активно размножающейся популяции всегда имеется некоторое количество лекарственно-устойчивых мутантов, которые практического значения не имеют, но по мере сокращения популяции, например под влиянием химиотерапевтических препаратов, изменяется соотношение между количеством лекарственно-чувствительных и лекарственно-устойчивых клеток. В этих условиях происходит размножение главным образом лекарственно-устойчивых клеток, их количество неуклонно возрастает. Итогом такой клональной селекции является поликлоновость опухоли и доминирование наиболее агрессивных клонов. В связи с этим целью настоящего обзора является обобщение данных по основным механизмам развития МЛУ опухолевых клеток при воздействии различных химиопрепаратов.
Аналитический обзор
1. Р-гликопротеин – основной белок-транспортер множественной лекарственной устойчивости
Р-гликопротеин – крупный трансмембранный белок с молекулярной массой 170 кДа (gp170, Pgp), состоящий из двух одинаковых частей, каждая из которых включает шесть гидрофобных трансмембранных участков (рис. 1). Это позволяет считать, что молекула Pgp 12 раз пересекает плазматическую мембрану клетки и имеет два сайта связывания с АТФ, свидетельствующих об энергозависимости функционирования белка (Chen et al., 1986; Roninson, 1991). Предполагается, что 12 трансмембранных доменов формируют поры или каналы, через которые Рgp активно выбрасывает вещества, и энергия гидролиза АТФ переносится от двух АТФ-связывающих доменов, обеспечивая работу выбрасывающего насоса (рис. 2) (Roninson, 1991).
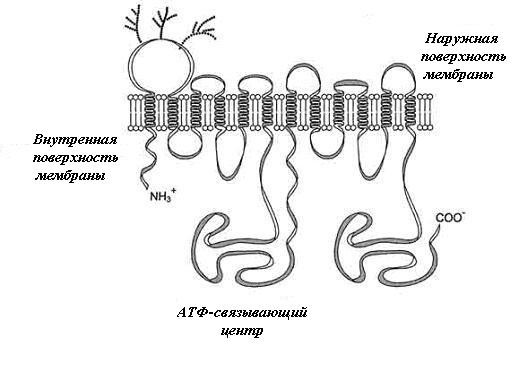
Рис. 1. Строение Р-гликопротеина
Р-гликопротеин – интегральный белок, имеющий 12 трансмембранных доменов, пронизывающих бислой цитоплазматической мембраны. N- и С-концы белка обращены в цитозоль. Участки Р-гликопротеина на наружной поверхности мембраны гликозилированы. Область между шестым и седьмым доменами имеет центры для присоединения АТФ и аутофосфорилирования (Биохимия, 2003)
Fig. 1. The structure of P-glycoprotein
P-glycoprotein is an integral membrane protein containing 12 transmembrane domains that span the plasma membrane bilayer. Protein N- and С- termini are exposed to the cytosol. The first extracellular loop of Р-glycoprotein is glycosylated. The region between the sixth and the seventh domains contains ATP-binding and autophosphorilation sites (Biochemistry, 2003)
Активность Р-гликопротеина определяет резистентность опухолевых клеток ко многим противоопухолевым лекарствам (антрациклиновым антибиотикам, алкалоидам растительного происхождения, в частности к винка-алкалоидам, подофиллотоксинам, таксанам и др.), а также ко многим другим веществам – флуоресцентным красителям (бромистому этидию – БЭ), пуромицину, грамицидину D и др. (Гринчук и др., 1996; Меликсетян и др., 1999).
Р-гликопротеин человека кодируется геном mdr1, принадлежащим к семейству mdr, локализованным в хромосоме 7. К МЛУ может приводить как изменение экспрессии гена mdr1, так и увеличение дозы гена – амплификация участка генома, содержащего ген mdr1, и еще пяти или шести сцепленных с ним генов (Borst, 1991; Невзглядова, Шварцман, 1992).
Активность Pgp может быть оценена по степени накопления меченного лекарства (например, 3Н-винбластина) или флуоресцирующего препарата (например, даунорубицина) клетками, а также по степени накопления или по скорости выброса веществ, например, флуоресцентных красителей, субстратов для Pgp (родамина-123) (Neyfakh, 1988; Гамалей и др., 1995).
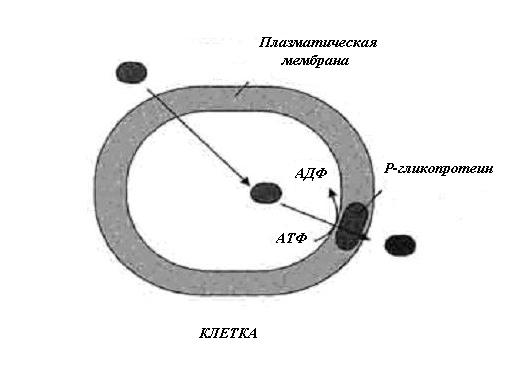
Рис. 2. Функционирование Р-гликопротеина
Темный овал – противоопухолевый агент (гидрофобное вещество) (Биохимия, 2003)
Fig. 2. The functioning of P-glycoprotein
An antitumor agent (hydrophobic compound) is depicted as the dark ellipse (Biochemistry, 2003)
С помощью гибридизации нуклеиновых кислот показано, что селекция клеток китайского хомячка CHLV-79 RJK на устойчивость к бромистому этидию приводит к амплификации и экспрессии генов семейства mdr, один из которых кодирует трансмембранный Р-гликопротеин, ответственный за понижение содержания БЭ в клетках. Авторы полагают, что амплифицированные копии генов mdr локализуются в тех же участках генома, где локализованы гены mdr дикого типа, и морфологически выявляются в виде гомогенно окрашенной области одной из хромосом, производной хромосомы 1. Полученные данные свидетельствуют о том, что устойчивость клеток CHLV-79 RJK к бромистому этидию в возрастающих концентрациях достигается в результате как амплификации генов mdr, так и регуляции экспрессии этих генов (Гринчук и др., 1993).
В другой работе в результате многоступенчатой селекции клеток китайского хомячка CHO-K1 на устойчивость к бромистому этидию были получены устойчивые сублинии Cerb-1 и Cerb-2. Показано, что клетки Сerb-1 и Cerb-2 характеризуются множественной лекарственной устойчивостью. С помощью гибридизации нуклеиновых кислот в клетках Cerb-2 обнаружены амплификация и повышенная экспрессия генов семейства mdr. В результате слияния клеток Cerb-2 с клетками китайского хомячка линии CHLV-79 RJK, чувствительными к БЭ, были получены два гибридных клона Hyrb-1 и Hyrb-2. Клетки Hyrb-2 характеризовались такой же устойчивостью к БЭ, как и клетки Cerb-2, у клеток Hybr-1 она оказалась ниже. В клетках Hyrb-2 амплификация и экспрессия генов mdr не отличались от таковых у клеток Cerb-2. В клетках Hyrb-1 амплификации этих генов не обнаружено, хотя уровень экспрессии оказался выше, чем в чувствительных клетках. На основании полученных данных можно предположить, что устойчивость клеток китайского хомячка к БЭ обусловлена амплификацией и повышенной экспрессией генов mdr (Липская и др., 1994).
В работе Гринчук и сотрудников (Гринчук и др., 1996) исследованы кариотипы клеток китайского хомячка линии CHLV-79 RJK и 6 сублиний этих клеток, отобранных на устойчивость к возрастающим концентрациям бромистого этидия, а также количество копий генов mdr в клетках, чувствительных и устойчивых к БЭ. Показано, что устойчивые к БЭ сублинии характеризовались МЛУ. Авторы предполагают, что амплификация генов mdr в устойчивых к БЭ клетках CHLV-79 RJK может рассматриваться как фактор, инициирующий последующую дестабилизацию генома и приводящий в итоге к изменениям в структуре кариотипа.
В настоящее время хорошо известно, что при ступенчатой селекции на устойчивость к некоторым цитотоксическим агентам (растительным алкалоидам и антибиотикам) клетки опухолевых линий in vitro приобретают перекрестную устойчивость к целому ряду цитостатиков, различающихся как по структуре и происхождению, так и по воздействию на разные клеточные мишени. Спектры препаратов, к которым развивается перекрестная устойчивость, а также механизмы, ее обеспечивающие, могут различаться в зависимости от выбора селективного агента.
В исследованиях Гринчук и сотрудников (Гринчук и др., 1998) в результате многошаговой селекции из популяции клеток хронического промиелолейкоза человека линии К562 были выделены три клона С9, В2 и В3, устойчивые к адриамицину (АДМ). Клетки клонов, устойчивые к АДМ, характеризовались перекрестной резистентностью к колхицину, актиномицину D и бромистому этидию – агентам группы МЛУ. При гибридизации ДНК по Саузерну в клетках устойчивых клонов была обнаружена амплификация гена mdr1. Кариологический анализ резистентных клеток, проведенный на ранних этапах культивирования в присутствии АДМ, показал наличие в геноме дополнительного генетического материала (морфологических маркеров амплификации – двойных мини-хромосом в клонах В2 и В3 и наличие гомогенно-окрашенных областей в хромосомах клеток клона С9). Выявленные кариотипические изменения рассматриваются как дестабилизация генома клеток К562/АДМ в связи с приобретением этими клетками фенотипа МЛУ.
Исследования генов и путей сигнальной трансдукции, вовлеченных в регуляцию активности Р-гликопротеина, свидетельствуют о том, что данных путей может быть несколько. Так, с использованием введения в клетку промоторной области гена mdr1 в составе конструкции, содержащей ген–репортер, а также в условиях стабильной трансфекции некоторых других генов было показано, что на активность промотора mdr1 или эндогенного гена клеток могут влиять гены р53, ras, raf и гены рецепторов ретиноевой кислоты (RARα и RARβ) (Teeter et al., 1991; Chin et al., 1992; Stromskaya et al., 1998). На активность гена mdr1 влияют также гены транскрипционных факторов c-fos и c-jun, для которых имеются респонсивные элементы в промоторной области гена mdr1.
В регуляции активности Р-гликопротеина принимают участие протеинкиназа С, протеинкиназа А и, возможно, еще некоторые протеинкиназы. Так, в работе Некрасовой (Некрасова, 1993) изучалась активность протеинкиназы С в клетках клонов линии CHO-K1, устойчивых к бромистому этидию в концентрациях 1 и 10 мкг/мл. Было обнаружено, что в клетках устойчивых клонов, отличающихся по пролиферативным характеристикам как друг от друга, так и от клеток исходной популяции, активность протеинкиназы С на первом этапе селекции увеличивалась во фракции мембран, а при дальнейшем увеличении устойчивости возрастала как в мембранах, так и в цитозоле. Культивирование устойчивых клеток в присутствии бромистого этидия приводило к увеличению активности фермента.
В опытах с культивируемыми клетками было показано, что процесс, связанный с развитием и становлением МЛУ, может сопровождаться изменениями в клеточной морфологии и физиологии: в частности, изменяются распластываемость клеток, скорость их размножения, внутриклеточный мембранный транспорт, опухолеродность и метастатическая активность резистентных клеток (Анисимов и др., 2003; Biedler, 1994). В работе Ерохиной и соавторов (Ерохина и др., 1997) описаны морфофункциональные изменения клеточных структур, сопровождающие первый этап возникновения лекарственной устойчивости в опухолевых клетках, экспрессирующих Р-гликопротеин. Были использованы фибробласты сирийского хомячка, трансформированные вирусом саркомы Рауса. Резистентные клетки получены путем культивирования в среде, содержащей субстрат для Рgp колхицин. При этом происходили изменения актинового цитоскелета, микротрубочек, аппарата Гольджи, везикулярного транспорта, сопровождающих развитие МЛУ.
Во многих работах установлена корреляция между дифференцировкой и экспрессией генов, кодирующих Р-гликопротеин в ответ на химиотерапевтические агенты. Так, Маркс с сотрудниками (Marks et al., 1993) показали, что экспрессия Р-гликопротеина в клетках К562, резистентных к винбластину и эпирубицину, не связана с экспрессией антигена CD34 и со специфическими маркерами эритроидной и миелоидной дифференцировки. В резистентных клетках отмечался более высокий уровень эритроидной и миелоидной дифференцировок, по сравнению с родительской линией.
В другой работе также было показано, что повышенный уровень экспрессии Р-гликопротеин-мРНК в клетках К562, обработанных бутиратом натрия, не связан с эритроидной дифференцировкой (Shibata et al., 1990). После обработки бутиратом натрия наблюдалось дозо-зависимое увеличение экспрессии Р-гликопротеин-мРНК в клетках К562, резистентных к адриамицину и винкристину. Гемин и митомицин С не влияли на уровень экспрессии Р-гликопротеин-мРНК. Авторы предполагают, что увеличение экспрессии Р-гликопротеин-мРНК, вероятно, может приводить к дифференцировке клеток К562.
Показано, что бутират натрия, гемин, 1-β-D-арабинофуранозилцитозин и эритроидный фактор дифференцировки (EDF) индуцируют эритроидную дифференцировку как в резистентных к винкристину клетках К562, так и клетках родительской линии. Уровень экспрессии mdr-гена в сублинии уменьшался после обработки EDF, но не после воздействия других индукторов. Следовательно, EDF регулирует функцию Р-гликопротеина в винкристин-резистентных клетках. Более того, EDF повышал чувствительность клеток сублинии к агентам множественной лекарственной устойчивости и подавлял экспрессию Р-гликопротеина (Okabe-Kado et al., 1991). Было также показано, что линия К562, резистентная к винбластину, была кросс-резистентна к доксорубицину и этопозиду, но оставалась чувствительной к цитозинарабинозиду, 6-тиохинолину. При этом MDR-фенотип не влиял на уровень синтеза гемоглобина в ответ на гемин, но увеличивал способность клеток дифференцироваться при обработке цитозинарабинозидом (Hait et al., 1993). Полученные результаты свидетельствуют, что индуцированная дифференцировка необходима в преодолении MDR-фенотипа.
Обработка клеток эритромиелолейкозной линии К562 адриамицином, алкациномицином и гемином приводит к увеличению синтеза гемоглобина. Такой тип дифференцировки связан с изменением экспрессии трех мембранных антигенов: GPA, CD15 и TfR. Кроме того, показано, что в сублинии клеток К562, резистентной к адриамицину, также наблюдается дифференцировка в ответ на отмеченные выше индукторы. Однако только адриамицин и алкациномицин влияли на экспрессию этих трех указанных антигенов, в то время как гемин не оказывал подобного действия (Oum’hamed et al., 1993).
Используя линию К562 и ее резистентную сублинию К562/E15B, была исследована дифференцировка этих клеток по двум направлениям: мегакариоцитарному – в ответ на обработку 12-О-тетрадеканоилфорбол-13-ацетатом (TPA) и эритроидному – после инкубации с бутиратом натрия. Обработка резистентной линии К562/E15B обоими типами индукторов приводила к появлению дифференцированного фенотипа, несмотря на повышенную экспрессию Р-гликопротеина. Причем при обработке клеток форболовым эфиром имела место повышенная экспрессия Р-гликопротеина, и, как следствие, происходило увеличение их резистентности и уменьшение накопления родамина-123 в этих клетках. В то время как для бутирата натрия подобного эффекта не наблюдалось. Таким образом, обработка резистентной линии форболовым эфиром приводит к изменению не только экспрессии Р-гликопротеина, но и его функциональной активности. Изложенные результаты подчеркивают важность изучения как экспрессии Р-гликопротеина в опухолевых клетках, так и функции этого белка при обработке дифференцирующими агентами (Marks et al., 1995).
Известно, что интерлейкин-1 и TNFα в комбинации с доксорубицином (DXR) усиливают антипролиферативную активность в доксорубицин-чувствительных (В16 меланома, Friend, K562) и резистентных к доксорубицину клеточных линиях (B16-DXR, FLC-DXR, K562-DXR) in vitro. При этом экспрессия mdr-генов приводит к сверхэкспрессии Р-гликопротеина и кросс-резистентности к винкристину. TNFα обладал большей антипролиферативной активностью в резистентных клетках В16 и Friend по сравнению с их родительскими вариантами. Комбинация TNFα и DXR приводила к синергическому ингибированию роста в родительских линиях В16 и К562, и частично в MDR-сублиниях этих клеток. К тому же TNFα и DXR индуцировали эритроидную дифференцировку в клетках К562, а в сублинии К562/DXR, кроме эритроидной, миелоидную дифференцировку. Авторы полагают, что комбинация DXR и TNFα необходима в преодолении MDR-фенотипа (Borsellino et al., 1994).
Анализ имеющихся в литературе данных, касающийся взаимосвязи индуцированной дифференцировки клеток опухолевых линий и индукции МЛУ, демонстрирует противоречивую картину. Как уже неоднократно отмечалось, индуцированная дифференцировка может сопровождаться экспрессией гена mdr1 и повышением активности Р-гликопротеина (Borsellino et al., 1994; Marks et al., 1993, 1995). Данные других авторов, основанные на экспериментах in vivo человека, предполагают отсутствие достоверной взаимосвязи между уровнем экспрессии гена mdr1 и степенью дифференцировки опухолевых клеток (Wang, Cai, 1998). Более того, с использованием клеток острого миелоидного лейкоза (ОМЛ) было показано, что менее зрелые клетки в образцах ОМЛ имеют пониженное накопление даунорубицина и более высокий темп его выброса. Таким образом, дифференцированные клетки в образцах ОМЛ лучше накапливают и удерживают ксенобиотик (Knaust et al., 2000). Принципиально сходные результаты представлены в работе Римет и соавторов (Rimet et al., 1999). Клетки человеческой опухолевой линии HT29-D4 трансфецировали геном mdr1 и индуцировали резистентность при инкубации с колхицином. Показано, что клетки полученных резистентных сублиний в меньшей степени способны к индукции дифференцировки, по сравнению с чувствительными к колхицину клетками.
2. Белки MRP и LRP в развитии множественной лекарственной устойчивости
Помимо Pgp МЛУ может определяться белком MRP (белок, ассоциированный с МЛУ, или multidrug resistance associated protein). Этот белок с молекулярной массой 190 кДа обеспечивает резистентность опухолевых клеток примерно к тому же кругу противоопухолевых препаратов, что и Pgp, он также является АТФ-зависимым и принадлежит к семейству АВС-транспортеров (Loe et al., 1996).
Хотя белок MRP определяет устойчивость клеток практически к тем же веществам, что и Pgp, однако спектр кросс-резистентности может отличаться. Клетки, экспрессирующие MRP, обычно проявляют меньшую кросс-резистентность к таксолу, чем клетки с Pgp-МЛУ. Обнаружено также, что для функционирования MRP необходим глутатион клетки. Следовательно, этот белок является одним из транспортеров коньюгатов глутатиона (насосы, обозначаемые GS-X) (Deeley, Cole, 1997; Borst et al., 1997).
Невысокие базальные уровни экспрессии MRP были обнаружены во всех клетках периферической крови, независимо от их дифференцировки (Loe et al., 1996). Имеются сведения о том, что в случаях острого миелолейкоза, резистентных к лечению, экспрессия MRP выше, чем в случаях, где достигнута полная ремиссия. Более высокое количество случаев, в которых клетки экспрессировали повышенные количества MRP, обнаружено при хроническом лимфолейкозе (Ставровская, 2000).
В отличие от белков Pgp и MRP открытый в 1993 г. белок МЛУ LRP (lung-resistance-related-protein; молекулярная масса 110 кДа) с помощью антител обнаруживается не на клеточной мембране, а в цитоплазме. Его экспрессируют клетки тканей, которые подвергаются токсическим воздействиям. Белок LRP является мажорным белком специфических клеточных органелл, рибонуклеопротеиновых частиц «vaults». Предполагают, что они могут участвовать в транспорте субстратов из ядра в цитоплазму. В клетке белок LRP нередко ассоциирован с везикулами и лизосомами, что позволяет связывать его функцию с транспортом и их секвестрацией внутри везикул (Ставровская, 2000). Имеются данные, свидетельствующие, что экспрессия LRP может быть причиной МЛУ при остром миелолейкозе (Wang, Xiao, 2012).
Несмотря на то что клиническая значимость белков MRP и LRP подтверждена недостаточно, в настоящее время проводятся исследования по поиску ингибиторов этих белков. Одним из таких ингибиторов является генистеин. Было найдено, что генистеин подавляет накопление даунорубицина в клетках, гиперэкспрессирующих MRP, но не в клетках с гиперэкспрессией Pgp. Генистеин высокотоксичен и не применяется в клинической практике, однако он используется в тестах на функциональную активность MRP.
3. Система глутатиона и множественная лекарственная устойчивость
Глутатион (GSH) – трипептид, синтезируемый в организме из глутаминовой кислоты, цистеина и глицина (рис. 3). Важнейшими функциями глутатиона являются антиоксидантная, иммунопротекторная и детоксификационная. Нарушение регуляции метаболизма глутатиона может стать причиной возникновения злокачественных опухолей. Повышенное количество глутатиона обнаруживается в клеточных линиях, резистентных к алкилирующим соединениям (эмбихину, хлорбутину, мелфалану, циклофосфамиду и др.). Химические взаимодействия между глутатионом и алкилирующими соединениями катализируются группой ферментов глутатион-S-трансфераз (GST), разные изоформы которых, вероятно, взаимодействуют с разными препаратами, повышая степень детоксикации лекарств. Таким образом, активация этих ферментов может определять резистентность клеток к лекарствам (Ставровская, 2000; Wang, Xiao, 2012).
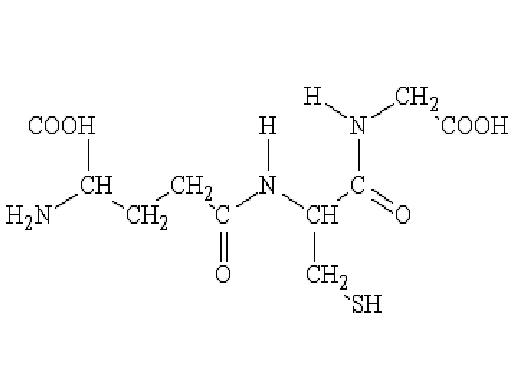
Рис. 3. Глутатион (γ-глутамилцистеинилглицин)
Fig. 3. Glutathione (γ-glutamyl-cysteinyl-glycine)
Было показано, что уровень GSH в клетках лейкоза Р388, резистентных к циклоплатаму (амин(цилопентиламин)-S-малатоплатине (II)), был почти в 10 раз выше, по сравнению с родительскими клетками этой линии. При этом активность глутатионпероксидазы и глутатионредуктазы была в 2 раза, а GST – в 1,5 раза выше в клетках резистентного штамма по сравнению с чувствительным. Введение циклоплатама мышам-опухоленосителям вызывало значительное увеличение уровня GSH в опухолевых клетках обоих штаммов, тогда как изменения в активности GSH-зависимых ферментов были выражены в меньшей степени. Полученные результаты свидетельствуют о том, что GSH-зависимые ферменты могут вносить существенный вклад в лекарственную устойчивость клеток лейкоза Р388 к циклоплатаму (Дедерер и др., 1995).
Ферменты, катализирующие синтез глутатиона в клетке, также могут иметь отношение к лекарственной устойчивости, однако их роль в МЛУ опухолевых клеток исследована недостаточно. С изменениями системы GSH может быть связана резистентность клеток не только к алкилирующим агентам, но и препаратам круга Pgp-МЛУ (антрациклинам и винкристину). Последнее указывает на то, что существуют общие механизмы регуляции mdr-генов и генов, участвующих в метаболизме глутатиона (Невзглядова, Шварцман, 1992).
Было показано, что в клеточной линии MCF7, резистентной к 4-нитрохинолин-1-оксиду (4-NQO) наблюдаются сверхэкспрессия Pgp и повышение активности GST. При этом такая синергетическая резистентность к 4-NQO связана с GST-зависимым увеличением образования 4-NQO-глутатион-коньюгатов, которые выводятся из клеток с помощью Pgp. Последнее необходимо для обеспечения полной защиты клеток от 4-NQO-токсичности (образования аддуктов с ДНК и цитотоксичности) (Morrow et al., 1998, 2000).
Таким образом, глутатион является компонентом для ряда ферментов, которые играют важную роль в защитной системе организма. Многие типы опухолей содержат много глутатиона, что позволяет злокачественным клеткам повысить сопротивляемость к химио- и радиотерапии. Тем не менее процессы регуляции метаболизма глутатиона в опухолевых клетках нарушены, и использование в данном случае лекарственных препаратов направленного действия в комбинации с цитостатиками может повысить эффективность антиопухолевой терапии.
4. Механизмы множественной лекарственной устойчивости, связанные с р53 и белками Bcl-2-семейства
В последние годы интенсивно исследуются механизмы возникновения МЛУ, связанные с подавлением апоптоза, индуцированного цитостатическими агентами. Показано, что нарушение регуляции генов, участвующих в контроле апоптоза (онкосупрессора р53 и генов семейства bcl-2), может приводить к приобретению клетками устойчивости к широкому спектру противоопухолевых препаратов – ДНК-тропным агентам (адриамицину, актиномицину D), антиметаболитам (5-фторурацилу), но не к соединениям, взаимодействующим с митотическим веретеном деления (колхицину, таксолу) (Miyashita, Reed, 1993; Hickman et al., 1994; Blagosklonny et al., 1997). В клинических исследованиях выявлена корреляция между уровнем экспрессии антиапоптотических генов bcl-2 и bcl-xL в различных опухолях и их высокой устойчивостью к химиотерапевтическим препаратам (Krajewska et al., 1996). На экспериментальных моделях показано, что отсутствие функционального гена bax, а также сверхэкспрессия генов bcl-2 и bcl-xLмогут приводить к возникновению МЛУ (Minn et al., 1995; McCurrach et al., 1996). Описаны также клеточные линии, фенотип МЛУ которых обусловлен сразу несколькими из вышеупомянутых механизмов, то есть имеет мультифакториальную природу.
Как было отмечено выше, важнейшими элементами ответа клетки на стрессорные воздействия, в том числе на воздействие химиотерапевтических препаратов, являются ген р53 и регулируемые им гены (Копнин, 2000; Чумаков, 2000). В норме ген р53 активируется в ответ на различные повреждающие клетку воздействия, что приводит к остановке клеточного цикла и/или апоптозу. В результате поврежденные клетки либо удаляются из популяции, либо у них появляется возможность репарировать поврежденную ДНК. Изменения в гене р53, весьма частые в опухолях, обуславливают нарушения его нормальной функции и нарушение способности клеток вступать в апоптоз или останавливаться в контрольных точках клеточного цикла в ответ на повреждение. Таким образом, нарушения функции р53 a priori могут привести к изменениям чувствительности опухолевых клеток к воздействию химиотерапевтических препаратов, в частности, к развитию множественной лекарственной устойчивости (Ставровская, 2000).
В число индукторов, активирующих нормальный р53, входят агенты, различным образом индуцирующие повреждения ДНК, например, через изменение пула нуклеотидов в клетке, изменение редокс-потенциала (в частности, при накоплении активного кислорода), разрушение веретена митотического деления и др. (Glaccia et al., 1998). Клетки с мутантным р53 чаще всего более резистентны к препаратам с разным механизмом действия (например, к цисплатину и 5-фторурацилу), чем клетки, содержащие в геноме дикий тип гена р53. Эти данные свидетельствуют о существенной роли р53 в определении чувствительности злокачественных новообразований к химиотерапии (Ставровская, 2000).
Исследования регуляции р53 показывают, что характер модификаций, активирующих этот белок в ответ на стимулы, зависит от стрессорного воздействия, от тканевой и видовой принадлежности клеток, от типа аминокислотной замены в мутантном р53 (Glaccia et al.,1998). Различные повреждающие агенты разными способами активируют р53, и эта активация осуществляется через разные сигнальные пути клетки. Например, активация р53 в ответ на повреждение ДНК приводит к гибели (апоптозу) фибробластов человека, в то время как размножение фибробластов грызунов лишь временно задерживается (Clarke et al., 1993). В одной из работ было обнаружено, что гиперэкспрессия р53 в культивируемых клетках лёгкого человека линии Н1299 преобладает. Однако цитотоксический эффект цисплатина защищает эти клетки от токсического действия випезида (Wang et al.,1996). Напротив, в других клетках (например, в тимоцитах мыши) р53 определяет индукцию апоптоза именно випезидом (Clarke et al.,1993).
Значительная корреляция между уровнем экспрессии р53 дикого типа и радиочувствительностью найдена при анализе клеточных линий опухолей человека, в частности лимфомы Беркитта и лимфобластом (Fan et al., 1994). Клеточные линии с высоким уровнем экспрессии р53 испытывали задержку в периоде G1 и были более радиочувствительны. Тимоциты, не синтезирующие р53, гораздо более резистентны к ионизирующим излучениям и этопозиду, чем р53-позитивные клетки, но сохраняют нормальную чувствительность к глюкокортикоидам и кальциевым ионофорам (Lowe et al., 1993; Clarke et al., 1993).
Показано, что инкубация клеток клона К562, несущих трансфецированный ген температурочувствительного мутанта р53 (K562/ts-p53), при 32 °С приводит к активации мутантного р53 и индукции апоптоза, который запускается, по-видимому, в ответ на предшествующие повреждения ДНК. Форболовый эфир способен понизить экспрессию р53 как в фибробластах, инкубированных с адриамицином, так и в клетках K562/ts-p53 (при инкубации последних при пермиссивной температуре 32 °С), что сопровождается подавлением апоптоза указанных клеток (Magnelli et al., 1995).
Белки, синтез которых контролирует транскрипционный фактор р53 (р21waf-1, MDM), также могут влиять на чувствительность клеток к цитостатикам. Факты, полученные в пользу их значения для лекарственной устойчивости опухолевых клеток, сходны с полученными для р53 (Ставровская, 2000).
Помимо опухолевых супрессоров на чувствительность клеток к различным цитостатическим агентам могут оказывать влияние антиапоптотические белки.
Так, показано влияние онкогена bcl-2 и других генов семейства bcl-2 на лекарственную устойчивость опухолевых клеток. Онкоген bcl-2 участвует в процессе возникновения злокачественного новообразования в связи с тем, что он подавляет апоптоз (Копнин, 2000). Ген bcl-2 принадлежит к большому семейству генов, продукты которых обладают как антиапоптотическим (например, Bcl-2, Bcl-XL), так и проапоптотическим действием (Bax, Bad) на клетки (Adams, Cory, 1998). Кодируемые этими генами полипептиды могут образовывать гомо- и гетеродимеры, которые в зависимости от составляющих компонентов определяют их влияние на апоптоз. Белок Bcl-2 способен тормозить апоптоз, вызываемый р53 в ответ на генотоксические воздействия. Показано, что гиперэкспрессия Bcl-2 сообщает клеткам лекарственную устойчивость к различным химиотерапевтическим препаратам (Dive, 1997). Существуют данные, показывающие, что экспрессия гена bcl-2 может рассматриваться как плохой прогностический признак при ряде новообразований, например, при раке мочевого пузыря, новообразованиях системы кроветворения лимфоидного и миелоидного рядов (Allouche et al., 1997).
Трансфекция различных клеточных линий bcl-2 предотвращает их гибель при воздействии облучения, многочисленных химиотерапевтических препаратов, ингибитора протеинкиназ стауроспорина, ингибитора топоизомеразы II этопозида, глюкокортикоидов и многих других. Однако Bcl-2 не подавляет гибель клеток-мишеней под влиянием цитотоксических лимфоцитов. Апоптоз, индуцированный через TNF и CD95, подавляется Bcl-2 в одних клеточных линиях и не ингибируется в других (Уманский, 1996).
Обнаружена интересная корреляция между уровнем Bcl-2, митохондриальной активностью и резистентностью к глюкокортикоидам различных линий лимфом человека и мыши (Smets et al.,1994). Более того, митохондриальные ингибиторы восстанавливают чувствительность резистентных клеточных линий к дексаметазону (Уманский, 1996).
Стабильная высокая экспрессия гена bcl-xL в клетках линий-трансфектантов К562/bcl-xL и HEL/bcl-xL сопровождается подавлением апоптоза указанных клеток, запускаемого индукторами эритроидной дифференцировки (гемином и ретиноевой кислотой). При этом трансфекция гена bcl-2 не затрагивает экспрессию маркеров эритроидной дифференцировки (в частности, ε-глобина) клеток в ответ на указанные индукторы (Benito et al., 1996).
Клетки человеческой гистоцитарной лимфомы U937 через 4 суток инкубации в присутствии блеомицина останавливаются в фазе G0/G1 клеточного цикла, приобретают признаки миелоидной дифференцировки (повышенную гранулярность и экспрессию CD11b) и подвергаются апоптозу. Инкубация клеток трансфектантов U937/bcl-2 в присутствии блеомицина в течение 2х недель сопровождается замедленной экспрессией маркеров миелоидной дифференцировки при отсутствии признаков апоптоза и блока пролиферации (Guedez, Zucali, 1996).
Повышенная экспрессия продуктов генов bcl-2 и bcl-xLв клетках линий трансфектантов К562/bcl-2 и К562/bcl-xL подавляет апоптоз, индуцированный ингибитором фосфопротеинфосфатаз РР1 и РР2А окадаевой кислотой (Benito et al., 1997). По отношению к клеткам К562, HL-60 и меланомы В16 окадаевая кислота выступает также как индуктор дифференцировки.
В работе Рэя с сотрудниками (Ray et al., 1996) модуляция экспрессии Bcl-xL-белка в клетках К562 достигалась путем трансфекции клеток родительской линии геном bcl-xS. Известно, что белок Bcl-xL в цитоплазме клеток может формировать комплекс с белком Bcl-xS, что приводит к снижению свободного белка Bcl-xL (Kharbanda et al., 1997). Показано, что повышенная экспрессия белка Bcl-xS в клетках-трансфектантах сопровождается усилением спонтанной и индуцированной (АраЦ и гексаметиленбисацетамид) эритроидной дифференцировки этих клеток и повышенной способностью к апоптозу в ответ на АраЦ (Ray et al., 1996). Усиление индуцированного апоптоза в ответ на этопозид и таксол наблюдается также в линии клеток-трансфектантов MCF-7/bcl-xS (Sumantran et al., 1995).
Ряд препаратов, применяемых при лечении злокачественных новообразований, являются ингибиторами топоизомераз (топоизомеразы I, топоизомеразы II (Топо II). В их круг входят соединения, стабилизирующие комплекс топоизомераза-ДНК, который в нормальных условиях быстро распадается. Фермент топоизомераза II является внутриклеточной мишенью ряда цитостатиков-антрациклинов, эпиподофиллотоксинов и аминоакридинов. Топоизомераза II играет важную роль при подготовке клеток к митозу, и, в частности, обеспечивает премитотическую конденсацию и сегрегацию хромосом. В клетках человека выявлены две формы Топо II – Топо IIα и Топо IIβ, кодируемые разными генами. α-изоформа белка более чувствительна к ингибированию цитостатиками, чем β-форма (van der Zee et al., 1994). В исследованиях клеточных линий, устойчивых к ингибиторам Топо II, выявлены снижение активности и (или) экспрессии Топо II, изменения в субклеточной локализации фермента и экспрессия мутантной формы белка, устойчивого к ингибированию цитостатиками. К группе препаратов, к которым возникает этот тип резистентности, принадлежат адриамицин, рубомицин, митоксантрон, этопозид и др. Таким образом, большинство веществ, взаимодействующих с молекулой топоизомеразы II, являются также субстратами Pgp и MRP.
Этопозид (VP-16) является ингибитором топоизомеразы II, тем самым вызывая повреждения ДНК, что в дальнейшем может привести к остановке клеточного цикла и/или апоптозу. Показано, что VP-16 может стимулировать c-jun и c-fos м-РНК экспрессию в некоторых клеточных линиях, в частности К562 и HL-60. VP-16 в концентрации 10 мкМ индуцирует межнуклеосомальную деградацию ДНК в клетках линии HL-60 через 6 часов инкубации, а в клетках К562 наблюдаются незначительные повреждения ДНК только после обработки 100 мкМ этопозида в течение 24 часов. Уровень экспрессии с-jun был одинаков в обеих клеточных линиях, однако синтез белка Bcl-2 в клетках HL-60 превышал в 13 раз аналогичный показатель в клетках К562 (Ritke, Yalowich, 1993).
При исследовании клеток хронического промиелолейкоза человека линии К562, устойчивых к этопозиду, были обнаружены снижение уровня экспрессии и активности ДНК-топоизомеразы II и увеличение экспрессии белка р85 по сравнению с клетками исходной линии К562. С другой стороны, скорость накопления этопозида в клетках устойчивого варианта была существенно снижена при отсутствии увеличения экспрессии гена mdr1. Авторы предположили существование в данном случае особого механизма устойчивости к этопозиду, не связанного с увеличением содержания Р-гликопротеина в клеточной мембране. Однако при исследовании клеток линии К562, устойчивых к адриамицину и обладающих фенотипом МЛУ, были выявлены повышенная экспрессия и амплификация гена mdr1 (Гринчук и др., 1998; Меликсетян и др., 1999). Очевидно, и этот тип МЛУ сосуществует иногда с другими механизмами лекарственной устойчивости в одних и тех же клетках.
Действие двух ингибиторов топоизомеразы II (амсакрина и доксорубицина) на экспрессию онкогена c-myc в клетках линии К562 и сублинии К562/DoxR было показано в работе Клари и сотрудников (Clary et al., 1998). Отмечено уменьшение уровня экспрессии c-myc после обработки обоими ингибиторами, с более выраженным эффектом после воздействия амсакрина на резистентную к доксорубицину линию. При этом амсакрин индуцировал апоптоз только в резистентной линии, в то время как к доксорубицину обе линии были резистентны. Результаты свидетельствуют, что онкоген c-myc не влияет на резистентность клеток К562/DoxR, но может активировать апоптотические пути в этих клетках, не индуцируя апоптоз в родительской линии.
При использовании в качестве ингибиторов топоизомеразы II камптотецина (СРТ) и этопозида (VP-16) было обнаружено, что кинетика фрагментации связана с клеточной чувствительностью. В высоких концентрациях (выше ЕC50) уровень фрагментации ДНК в чувствительных линиях (BV173, HL-60, U937) сильно превышал этот уровень в резистентных к обоим соединениям линиях (К562, KCL22). Длительная экспозиция этопозида и камптотецина вызывала остановку клеточного цикла либо в G2-, либо в S-периоде, что было связано как с клеточной чувствительностью, так и с высокими концентрациями использованных соединений (Dublez et al., 1995).
Таким образом, как анти-, так и проапоптотические белки могут влиять на развитие резистентности опухолевых клеток. Степень индукции такой резистентности зависит от гистогенетической принадлежности клеток.
5. Филадельфийская хромосома как одна из причин развития множественной лекарственной устойчивости
Наряду с продуктами генов bcl-2 и bcl-x способностью ингибировать апоптоз клеток, вызванный обработкой дифференцирующими агентами, обладает гибридный белок BCR/ABL. Данный белок является продуктом химерного гена, расположенного в аномальной хромосоме (филадельфийской хромосоме – Ph). Хромосома была впервые открыта и описана в 1960 г. учёными из Филадельфии (Пенсильвания, США): Питером Ноуеллом (Пенсильванский университет) и Дэвидом Хангерфордом (университет Темпл). Филадельфийская хромосома образуется вследствие реципрокной транслокации материала длинных плечей хромосом 9 и 22 — t (9;22) (q34;qll) (рис. 4). В результате, к части BCR-гена из хромосомы 22 прикрепляется ABL-ген из хромосомы 9. Этот аномальный «слившийся» ген генерирует белок p210 или, иногда, p185. Белок является тирозин-киназой.
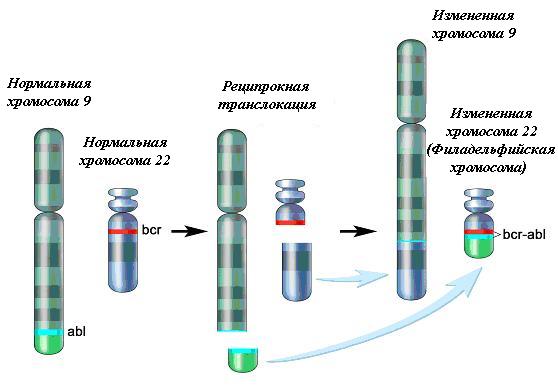
Рис. 4. Образование филадельфийской хромосомы (Ph-хромосомы)
Участок хромосомы 9 и участок хромосомы 22 отрываются и меняются местами (Острый лимфобластный лейкоз ..., 2012 )
Fig. 4. The Philadelphia chromosome (Ph-chromosome) formation
A piece of chromosome 9 and a piece of chromosome 22 break off and switch places with each other (Acute lymphoblastic leukemia..., 2012 )
Во многих случаях проявления хронического миелолейкоза и острого лимфобластного лейкоза связаны с экспрессией BCR/ABL-белка. Так, показано, что клетки К562 обычно являются резистентными к апоптотической индукции, вследствие повышенной экспрессии этого химерного белка. Однако другие исследователи, изучая воздействие ингибиторов топоизомеразы II (амсакрина и доксорубицина), обнаружили, что амсакрин, и в меньшей степени доксорубицин, могут индуцировать апоптоз в доксорубицинрезистентных вариантах этой линии. Было также показано, что амсакрин в невысоких концентрациях способен уменьшать экспрессию bcr/abl на 20 % только в линии, резистентной к доксорубицину, в то время как для доксорубицина и этопозида этого эффекта не наблюдалось. Амсакрин не влиял на экспрессию bcl-xLв резистентной линии. Таким образом, авторы подчеркивают роль bcr/abl в защите клеток против апоптоза и возможное использование специфических топоизомеразных ингибиторов в преодолении резистентности к апоптозу (Laroche-Clary et al., 2000).
Показано, что идарубицин индуцирует экспрессию Bcl-2 в некоторых резистентных лейкемических клеточных линиях (U937, HL-60, K562), которая может ингибироваться циклогексимидом и актиномицином D. Синтез Bcl-2 приводит к увеличению отношения Bcl-2/Bax только в линиях, резистентных к апоптотической индукции. При этом было замечено, что отношение Bcl-2/Bax отражает апоптотическую способность лучше, чем постоянно высокий уровень экспрессии Bcl-2 в этих клетках (Durrieu et al., 1999).
Исследования, проведенные Доу и сотрудниками (Dou et al., 1999), показали, что протеосомы могут играть роль в регуляции функции Bcr-Abl. Ингибирование протеосомальной активности, но не активности цистеиновых протеаз, приводит к запуску апоптотической программы в клетках линии К562. Это связано с тем, что протеосомальный ингибитор приводит к значительному уменьшению экспрессии онкопротеина Bcr/Abl, однако уровни c-Abl/p145 и Bcr/p160 оставались неизменны. Полученные результаты свидетельствуют, что ингибирование протеосомальной активности является достаточным для инактивации функций Bcr/Abl и активации апоптоза в клетках, резистентных к химиотерапии.
Тирозинкиназная активность онкогена bcr/аbl приводит к трансформации гематопоэтических клеток. Показано, что тирозинкиназный ингибитор STI571 ингибирует BCR/ABL, TEL/ABL, v-ABL-киназную активность и снижает жизнеспособность клеток, трансформированных одним из этих онкогенов. В дальнейшем исследователями были получены две BCR/ABL-позитивные клеточные линии (Ba/F3 и К562), обладавшие частичной резистентностью к STI571. В резистентной линии Ba/F3.p210 была выявлена амплификация гена BCR/ABL, в отличие от линии К562, хотя в обеих клеточных линиях отмечено значительное увеличение экспрессии р210 BCR/ABL белка. Добавление STI571 к обеим резистентным линиям вызывает значительное уменьшение фосфорилирования клеточных белков по тирозину, что также было характерно и для чувствительных клеток. Однако ингибирование киназной активности было временным и частичным и не было связано с апоптотическим процессом. Таким образом, авторы подчеркивают, что резистентность к STI571 может иметь мультифакториальную природу (Weisberg, Griffin, 2000).
Несмотря на то что многие сигналинговые молекулы могут активироваться Bcr/Abl-киназой, антиапоптотический путь, запускаемый онкогеном bcr/abl, остается до конца не выясненным. Хорита и сотрудники (Horita et al., 2000) показали, что интерлейкин-3-зависимая экспрессия антиапоптотического белка Bcl-xL индуцируется Bcr/Abl посредством активации STAT5 (signal transducer and activator of transcription). Ингибирование Bcr/Abl-киназной активности в Bcr/Abl-экспрессирующих и CD34(+)-клетках больных хроническим миелоидным лейкозом приводит к апоптозу, посредством подавления способности STAT5 взаимодействовать с bcl-x-промотором. Было замечено, что после ингибирования Bcr/Abl-киназы, экспрессия Bcl-xL подавляется в хронической фазе быстрее, чем при бластном кризе. Последнее позволяет предположить, что это один из новых антиапоптотических путей, запускаемый Bcr/Abl, и модуляция этого пути, возможно, позволит исследователям преодолеть резистентность лейкозных клеток к некоторым химиотерапевтическим препаратам.
Аденовирус Е1А может индуцировать апоптоз в BCR/ABL-экспрессирующих клетках и LAMA-84 лейкемических клетках. Такая апоптотическая активность Е1А связана с активацией каспазы-3 и поли(АДФ-рибозо)полимеразы. Благодаря трансфекции аденовируса Е1А, клетки К562, резистентные к химиотерапии, становятся чувствительными к этопозиду и даунорубицину. Следовательно, использование аденовируса Е1А в комбинации с некоторыми химиотерапевтическими агентами можно рассматривать как перспективное направление для успешного лечения Bcr/Abl-экспрессирующих лейкозов (Stiewe et al., 2000).
Наряду с высокой экспрессией Bcr/Abl на резистентность клеток может оказывать влияние и активность протеинкиназы С (ПКС), в частности для резистентности клеток К562 к таксол-индуцированному апоптозу. Обработка этих клеток таксолом приводит к увеличению активности ПКС. Напротив, Bcr/Abl-негативные миелолейкозные клетки HL-60 были чувствительны к таксол-индуцируемому апоптозу, и в них не наблюдалась активация ПКС. Обработка клеток К562 AG957 – специфическим ингибитором Bcr/Abl блокирует активацию ПКС, вызванную таксолом, и увеличивает их чувствительность к апоптозу (Jamieson et al., 1999).
В работе Канг и сотрудников (Kang et al., 2000) показано, что апоптоз может индуцироваться после обработки некоторых клеточных линий PD98059 специфическим ингибитором MEK-киназ (MAPK и ERK-киназа (extracellular signal-regulated kinase)), но не после обработки сорбитолом. Сорбитол, как известно, является сильным активатором SAPK (stress-activated protein kinase) и р38-киназы. Было обнаружено, что клетки К562 резистентны к сорбитолиндуцированному апоптозу, в то время как в линиях Jurkat, HL-60 и U937 после воздействия сорбитола отмечалась фрагментация ДНК. При этом апоптозчувствительные клетки имели низкую активность JNK/SAPK и р38-киназы, сходную с активностью этих киназ в клетках К562 после обработки сорбитолом. Клетки К562 имеют высокий базальный уровень активности ERK/MAPK, по сравнению с другими, чувствительными к апоптозу, клеточными линиями. В дальнейшем в клетках линии HL-60 индуцированный сорбитолом апоптоз был предотвращен обработкой форбол-12-миристат-13-ацетатом, который активирует ERK/MAPK-путь. Авторы полагают, что ингибирование ERK/MAPK-пути, но не активация JNK/SAPK, способствует индукции апоптоза в клетках К562. Вместе с тем так же, как и в случаях других механизмов, характер влияния данной системы на лекарственную чувствительность опухолевых клеток не всегда однозначен и, очевидно, зависит от целого ряда параметров.
Заключение
Изложенные выше результаты изучения возникновения и формирования механизмов МЛУ опухолевых клеток показывают, что эти механизмы множественны. Установление новых генов и белков, обуславливающих защиту клетки от повреждений, а также изучение сигналинговых путей клетки, в которые вовлечены эти белки, открывают новые перспективы для исследования существа механизмов МЛУ, разнообразие которых значительно затрудняет как диагностику причин устойчивости больных к химиотерапии, так и выработку разумных способов преодоления МЛУ. Тем не менее современные методы направленного синтеза химических реагентов, используемых в онкологической практике, в совокупности с методами направленной доставки химиопрепаратов в конкретный клеточный компартмент позволят в ближайшем будущем решить вопрос, связанный с преодолением МЛУ.
Библиография
Анисимов А. Г., Чекмасова А. А.,. Волкова Т. О., Немова Н. Н. Эритроидная дифференцировка сублиний клеток К562, резистентных к 2-(4’-диметил-аминостирил)хинолин-1-оксиду или 4-нитрохинолин-1-оксиду, значительно усиливается при обработке тимидином // Известия РАН. Сер. биол. 2003. № 1. С. 37-46.
Гамалей И. А., Березкина Е. В., Ковалева З. В., Игнатова Т. Н. Кинетика выхода родамина 123 из клеток с множественной лекарственной устойчивостью при действии ингибиторов энергетического метаболизма // Цитология. 1995. Т. 37. № 1/2. С. 118-125.
Биохимия: Учеб. для вузов, Под ред. Е.С. Северина. 2003. 779 с. URL: http://www.biochemistry.ru/biohimija_severina/B5873Part95-617.html (дата обращения 20.05.2012)
Гринчук Т. М., Липская Л. А., Арцыбашева И. В., Сорокина Е. А., Паньшина Ю. Т., Игнатова Т. Н. Изменчивость кариотипа клеток китайского хомячка CHLV-79 RJK, характеризующихся множественной лекарственной устойчивостью, обусловленная амплификацией генов семейства MDR // Цитология. 1996. Т. 38. № 2. С. 161-171.
Гринчук Т. М., Липская Л. А., Васюхин В. И., Сорокина Е. А., Арцыбашева И. В., Игнатова Т. Н. Амплификация и сверхэкспрессия генов mdr в клетках китайского хомячка CHLV-79 RJK, устойчивых к бромистому этидию, сочетается с наличием кариотипических маркеров амплификации // Цитология. 1993. Т. 35. № 6/7. С. 86-97.
Гринчук Т. М., Павленко М. А., Липская Л. А., Сорокина Е. А., Тарунина М. В., Березкина Е. В., Ковалева З. В., Игнатова Т. Н. Устойчивость клеток хронического промиелолейкоза человека линии К562 к адриамицину коррелирует с направленной дестабилизацией генома – амплификацией гена MDR1 и неслучайными изменениями в структуре кариотипа // Цитология. 1998. Т. 40. № 7. С. 652-660.
Дедерер Л. Ю., Ланкин В. З., Коновалова А. Л., Горбачева Л. Б. Исследование биохимических механизмов резистентности к новому противоопухолевому препарату амин(циклопентиламин)-S-(-)-малатоплатине (II) (циклоплатаму) // Биохимия. 1995. Т. 60. № 4. С. 602-609.
Ерохина М. А., Ставровская А. А., Онищенко Г. Е. Реорганизация элементов цитоскелета и вакуолярной системы в опухолевых клетках на ранних этапах развития множественной лекарственной устойчивости // Цитология. 1997. Т. 39. № 11. С. 1038-1045.
Копнин Б. П. Мишени действия онкогенов и опухолевых супрессоров: Ключ к пониманию базовых механизмов канцерогенеза // Биохимия. 2000. Т. 65. № 1. С. 5-33.
Липская Л. А., Гринчук Т. М., Ефимова Е. В., Арцыбашева И. В., Сорокина Е. А., Васюхин В. И., Игнатова Т. Н. Амплификация и сверхэкспрессия генов семейства MDR в устойчивых к бромистому этидию клетках китайского хомячка СНО-К1 и в гибридах чувствительных и устойчивых клеток // Цитология. 1994. Т. 36. № 12. С. 1236-1244.
Меликсетян М. Б., Березкина Е. В., Павленко М. А., Гринчук Т. М. Исследование механизмов лекарственной устойчивости двух клеточных линий хронического промиелолейкоза человека линии К562, резистентных к ингибиторам ДНК-топоизомеразы II адриамицину и этопозиду // Цитология. 1999. Т. 41. № 7. С. 615-622.
Невзглядова О. В., Шварцман П. Я. Множественная устойчивость эукариотических клеток, обусловленная Р-гликопротеином // Молекуляр. биол. 1992. Т. 26. № 3. С. 487-499.
Некрасова Т. П. Различия в активности протеинкиназы С в клетках СНО-К1 и в клетках клонов этой линии, устойчивых к бромистому этидию // Цитология. 1993. Т. 35. № 8. С. 38-45.
Острый лимфобластный лейкоз у детей – Обзор. (Acute lymphoblastic leukemia in children. Overview.) URL: http://kyev-web.at.ua/index/ostryj_limfoblastnyj_lejkoz_u_detej_obzor/0-99 (дата обращения 20.5.2012)
Ставровская А. А. Клеточные механизмы множественной лекарственной устойчивости опухолевых клеток // Биохимия. 2000. Т. 65. № 1. С. 112-126.
Уманский С. Р. Апоптоз: молекулярные и клеточные механизмы // Молекуляр. биол. 1996. Т. 30. № 3. С. 487-502.
Чумаков П. М. Функция гена р53: выбор между жизнью и смертью // Биохимия. 2000. Т. 65. № 1. С. 34-47.
Adams J. M., Cory S. The Bcl-2 protein family: arbiters of cell survival // Science. 1998. Vol. 281. P. 1322-1326.
Allouche M., Bettaieb A., Vindis C., Rousse A., Grignon C., Laurent G. Influence of the Bcl-2 overexpression on the ceramide pathway in daunorubicin-induced apoptosis of leukemic cells // Oncogene. 1997. Vol. 14. № 15. P. 1837-1845.
Benito A., Lerga A., Silva M., Leon J., Fernandez-Luna J. L. Apoptosis of human myeloid leukemia cells induced by an inhibitor of protein phosphatases (okadaic acid) is presented by Bcl-2 and Bcl-X(L) // Leukemia. 1997. Vol. 11. P. 940-944.
Benito A., Silva M., Grillot D., Nuñez G., Fernández-Luna J. L. Apoptosis induced by erythroid differentiation of human leukemia cell lines is inhibited by Bcl-XL // Blood. 1996. Vol. 87. P. 3837-3843.
Biedler J.L. Drug resistance: genotype versus phenotype – thirty-second G. H. A. Clowes memorial award lecture // Cancer Res. 1994. Vol. 54. P. 666-678.
Blagosklonny M. V., Giannakakou P., El-Deiry W.-V., Kingston D. G., Higgs P. I., Neckers L., Fojo T. Raf-1/bcl-2 phosphorylation: a step from microtubule damage to cell death // Cancer Res. 1997. Vol. 57. P. 130-135.
Borsellino N., Crescimanno M., Flandina C., Flugy A., D'Alessandro N. Combined activity of interleukin-1 alpha or TNF-alpha and doxorubicin on multidrug resistant cell lines: evidence that TNF and DXR have synergistic antitumor and differentiation-inducing effects // Anticancer Res. 1994. Vol. 14. P. 2643-2648.
Borst P. Genetic mechanisms of drug resistance. A review // Acta Oncol. 1991. Vol. 30. P. 87-101.
Borst P., Kool M., Evers R. Do cMOAT (MRP2), other MRP homologues, and LRP play a role in MDR? // Sem. Cancer Biol. 1997. Vol. 8. P. 205-213.
Chen C. J., Chin J. E., Ueda K., Clark D. P., Pastan I., Gottesman M. M., Roninson I. B. Internal duplication and homology with bacterial transport prоteins in the mdrI (P-glycoprotein) gene from multidrug- resistant human cells. // Cell. 1986. Vol. 47. P. 381-389.
Chin K-V., Ueda K., Pastan I., Gottesman M. M. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53 // Science. 1992. Vol. 255. P. 459-462.
Clarke A. R., Purdie C. A., Harrison D. J., Morris R. G., Bird C. C., Hooper M. L., Wyllie A. H. Thymocyte aapoptosis induced by p53-dependent and independent pathways // Nature. 1993. Vol. 362. P. 849-858.
Clary A., Larrue A., Pourquier P., Robert J. Transcriptional down-regulation of c-myc expression in an erythroleukemic cell line, K562, and its doxorubicin-resistant variant by two topoisomerase II inhibitors, doxorubicin and amsacrine // Anticancer Drugs. 1998. Vol. 9. P. 245-254.
Deeley R., Cole S. Function, evolution and structure of multidrug resistance protein (MRP) // Sem. Cancer Biol. 1997. Vol. 8. P. 193-204.
Dive C. Avoidance of apoptosis as a mechanism of drug resistance // J. Intern. Med. 1997. Vol. 242. P. 139-145.
Dou Q. P., McGuire T. F., Peng Y., An B. Proteasome inhibition leads to significant reduction of Bcr-Abl expression and subsequent induction of apoptosis in K562 human chronic myelogenous leukemia cells // J. Pharmacol Exp. Ther. 1999. Vol. 289. № 2. P. 781-790.
Dublez L., Goldwasser F., Genne P., Pommier Y., Solary E. The role of cell cycle regulation and apoptosis triggering in determining the sensitivity of leukemia cells to topoisomerase I and II inhibitors // Leukemia. 1995. Vol. 9. № 6. P. 1013-1024.
Durrieu F., Belaud-Rotureau M. A., Lacombe F., Dumain P., Reiffers J., Boisseau M. R., Bernard P., Belloc F. Synthesis of Bcl-2 in response to anthracycline treatment may contribute to an apoptosis-resistant phenotype in leukemic cell lines // Cytometry. 1999. Vol. 36. № 2. P. 140-149.
Glaccia A. J., Kastan M. B. The complexity of p53 modulation: emerging patterns from divergent // Genes. Dev. 1998. Vol. 12. P. 2973-2983.
Guedez L., Zucali J. Bleomycin-induced differentiation of bcl-2-transfected U937 leukemia cells // Cell Growth Differ. 1996. Vol. 7. P. 1625-1631.
Fan S., El-Deiry W. S., Bae I., Freeman J., Jondle D., Bhatia K., Fornace A. J. Jr., Magrath I., Kohn K. W., O'Connor P. M. p53 gene mutations are associated with decreased sensitivity of human lymphoma cells to DNA damaging // Cancer Res. 1994. Vol. 54. P. 5824-5830.
Jamieson L., Carpenter L., Biden T. J., Fields A. P. Protein kinase Ciota activity is necessary for Bcr-Abl-mediated resistance to drug-induced apoptosis // J. Biol. Chem. 1999. Vol. 274. № 7. P. 3927-3930.
Hait W. N., Choudhury S., Srimatkandada S., Murren J. R. Sensitivity of K562 human chronic myelogenous leukemia blast cells transfected with a human multidrug resistance cDNA to cytotoxic drugs and differetiating agents // J. Clin. Invest. 1993. Vol. 91. № 5. P. 2207-2215.
Hickman J. A., Potten C., Merritt J., Fisherr T. Apoptosis and cancer chemotherapy // Phil. Trans. R. Soc. 1994. Vol. 345. P. 319-325.
Horita M., Andreu E.J., Benito A., Arbona C., Sanz C., Benet I., Prosper F., Fernandez-Luna J. L. Blockade of the Bcr-Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5-dependent expression // J. Exp. Med. 2000. Vol. 191. № 6. P. 977-984.
Kang C. D., Yoo S. D., Hwang B. W., Kim K. W., Kim D. W., Kim C. M., Kim S. H., Chung B. S. The inhibition of ERK/MAPK not the activation of JNK/SAPK is primarily required to apoptosis in chronic myelogenous leukemic K562 cells // Leuk. Res. 2000. Vol. 24. № 6. P. 527-534.
Kharbanda S., Pandey P., Schofield L., Israels S., Roncinske R., Yoshida K., Bharti A., Yuan Z. M., Saxena S., Weichselbaum R., Nalin C., Kufe D. Role for Bcl-xL as an inhibitor of cytosolic cytochrome C accumulation in DNA damage-induced apoptosis // Proc Natl. Acad. Sci. USA. 1997. Vol. 94. P. 6939-6942.
Knaust E., Porwit-MacDonald A., Gruber A., Xu D., Peterson C. Heterogeneity of isolated mononuclear cells from patients with acute myeloid leukemia affects cellular accumulation and efflux of daunorubicin // Haematologica. 2000. Vol. 85. P. 124-132.
Krajewska M., Moss S. F., Kraewski S., Song K., Holt P. R., Reed J. C. Elevated expression of Bcl-x and reduced Bak expression in primary rectal adenocarcinomas // Cancer Res. 1996. Vol. 56. P. 2422-2432.
Laroche-Clary A., Larrue A., Robert J. Down-regulation of bcr-abl and bcl-x(L) expression in a leukemia cell line and its doxorubicin-resistant variant by topoisomerase II inhibitors // Biochem. Pharmacol. 2000. Vol. 60. № 12. P. 1823-1828.
Loe D. W., Deepley R. G., Cole S. P. Biology of the multidrug resistance-associated protein, MRP // Eur. J. Cancer. 1996. Vol. 32 A. P. 945-957.
Lowe S. W., Schmitt E. M., Smith S. W., Osborne B. A., Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes // Nature. 1993. Vol. 362. P. 847-849.
Magnelli L., Cinelli M., Chiarugi V. Phorbol esters attenuate the expression of the p53 in cells treated with doxorubicin and protect ts-p53/K562 from apoptosis // Biochem. Biophys. Res. Commun. 1995. Vol. 215. P. 641-645.
Marks D. C., Davey M. W., Davey R. A., Kidman A. D. Differentiation and multidrug resistance in response to drug treatment in the K562 human leukaemia cell line // Br. J. Haematol. 1993. Vol. 84. № 1. P. 83-89.
Marks D. C., Davey M. W., Davey R. A., Kidman A. D. Expression of multidrug resistance to differentiation in the K562 human leukaemia cell line // Biochem. Pharmacol. 1995. Vol. 50. № 4. P. 475-480.
McCurrach M. E., Connor T. M., Knudson M., Korsmeyer S. Bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis // Proc. Nat. Sci. USA. 1996. Vol. 94. P. 2345-2349.
Miyashita T., Reed J. C. Bcl-2 oncoprotein blocks chemotherapy induced apoptosis in a human leukemia cell line // Blood. 1993. Vol. 8. P. 151-157.
Minn A., Rudin C. M., Boise L. H. Expression of Bcl-xL can confer multidrug resistant phenotype // Blood. 1995. Vol. 86. P. 1903-1907.
Morrow C. S., Diah S., Smitherman P. K., Schneider E., Townsend A. J. Multidrug resistance protein and glutathion S-transferase P1-1 act in synergy to confer protection from 4-nitroquinoline 1-oxide toxicity // Carcinogenesis. 1998. Vol. 19. № 1. P. 109-115.
Morrow C. S., Smitherman P. K., Townsend A. J. Role of multidrug-resistance protein 2 in glutathione S-transferase P1-1-mediated resistance to 4-nitroquinoline 1-oxide toxicities in HepG2 cells // Mol. Carcinog. 2000. Vol. 29. № 3. P. 170-178.
Neyfakh A. A. Use of fluorescent dues as molecular probes for the study of multidrug resistance // Exp. Cell Res. 1988. Vol. 174. P. 168-176.
Okabe-Kado J., Hayashi M., Honma Y., Hozumi M., Tsuruo T. Inhibition by erythroid differentiation factor (activin A) of P-glycoprotein expression in multidrug-resistant human K562 erythroleukemia cells // Cancer Res. 1991. Vol. 51. № 10. P. 2582-2586.
Oum’hamed Z., Joly P., Broglio C., Dufer J., Desplaces A. Study of erythroid differentiation of K562 cells resistant to adriamycin // Ann. Pharm. Fr. 1993. Vol. 51. № 5. P. 239-249.
Ray S., Bullock G., Nunez G., Tang C., Ibrado A. M., Huang Y., Bhalla K. Enforced expression of Bcl-XS induced differentiation and sensitizes chronic myelogenous leukemia-blast crisis K562 cells to 1-beta-D-arabinofuranosylcytosine-mediated differentiation and apoptosis // Cell Growth Differ. 1996. Vol. 7. P. 1617-1623.
Rimet O., Mirrione A., Barra Y. Multidrug-resistant phenotype influences the differentiation of a human colon carcinoma cell line // Biochem. Biophys. Res. Commun. 1999. Vol. 259. P. 43-49.
Ritke M. K., Yalowich J. C. Altered gene expression in human leukemia K562 cells selected for resistance to etoposide // Biochem. Pharmacol. 1993. Vol. 46. № 11. P. 2007-2020.
Roninson I. B. Structure and evolution of P-glycoprotein. Plenum Press, New York. 1991. P. 189-209.
Shibata H., Kanamaru R., Sato T., Ishioka C., Konishi Y., Ishikawa A., Wakui A., Tsuruo T. Increase in the level of P-glycoprotein mRNA expression in multidrug-resistant K562 cell lines treated with sodium butyrate is not accompanied with erythroid differentiation // Jpn. J. Cancer Res. 1990. Vol. 81. № 12. P. 1214-1217.
Smets L. A., Van den Berg J., Acton D., Top B., Van Rooij H., Verwijs-Janssen M. BCL-2 expression and mitochondrial activity in leukemic cells with different sensitivity to glucocorticoid-induced apoptosis // Blood. 1994. Vol. 84. P. 1613-1619.
Stromskaya T. P., Rybalkina E. Y., Shtil A. A., Zabotina T. N., Filippova N. A., Stavrovskaya A. A. Influence of exogenous RAR alpha gene on MDR1 expression and P-glycoprotein function in human and rodent cell lines // Br. J. Cancer. 1998. Vol. 77. P. 1718-1725.
Stiewe T., Parssanedjad K., Esche H., Opalka B., Pützer B. M. E1A overcomes the apoptosis block in BCR-ABL + leukemia cells and renders cells susceptible to induction of apoptosis by chemotherapeutic agents // Cancer Res. 2000. Vol. 60. № 14. P. 3957-3964.
Sumantran V. N., Ealovega M. W., Nunez G., Clarke M. F., Wicha M. S. Overexpression of Bcl-XS sensitizes MCF-7 cells to chemotherapy-induced apoptosis // Cancer Res. 1995. Vol. 55. P. 2507-2510.
Teeter L. D., Ecksberg T., Tsai Y., Kuo M. T. Analysis of the chinese hamster P-glycoprotein-multidrug resistance gene pgp1 reveals that the AP-1 site is essential for full promoter activity // Cell Growth Differ. 1991. Vol. 2. P.429-437.
van der Zee A. G. J., De Jong S., Keith W. N., Hollema H., Boonstra H., de Vries E. G. Quantitative and qualitative aspects of topoisomerase I and topoisomerase IIα and IIβ in untreated and platinum/cyclophosphamide treated malignant ovarian tumours // Cancer Res. 1994. Vol. 54. P. 749-755.
Volkova T. O., Bagina U. S., Zykina N. S., Malysheva I. E., Poltorak A. N. Treatment of cells К562/4-NQO and К562/2-DQO with chemical compounds of multidrug resistance leads to apoptosis / Bioinformatics of Genome Regulation and Structure / Systems of Biology – BGRS/SB-2010. Novosibirsk, 2012. Р. 40-42.
Wang S., Cai G. Clinical study of multi-drug resistance gene (MDR1) expression in primary ovarian cells // J. Tongji. Med. Univ. 1998. Vol. 18. P. 58-60.
Wang J., Xiao Z. RQ-PCR detection of GST-π and LRP genes in adult acute leukemia and its clinical significance // Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2012. Vol. 20. № 1. P. 78-82.
Wang Q., Fan S., Eastman A., Worland P. J., Sausville E. A., O'Connor P. M. UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53 // J. Nat. Cancer Inst. 1996. Vol. 88. № 14. Р. 956-965.
Weisberg E., Griffin J. D. Mechanism of resistance to the ABL tyrosine kinase inhibitor STI571 in BCR/ABL-transformed hematopoietic cell lines // Blood. 2000. Vol. 95. № 11. P. 3498-3505.
Благодарности
Работа выполнена при финансовой поддержке гранта Правительства РФ (Постановление № 220), ГК № 11.G34.31.0052 (ведущий ученый - профессор А. Н. Полторак).

 © 2011 - 2026
© 2011 - 2026